 CAP TODAY Pathology/Laboratory Medicine/Laboratory Management
CAP TODAY Pathology/Laboratory Medicine/Laboratory Management

William Check, PhD
April 2016—With next-generation sequencing’s clear benefits—for diagnosis, prognosis, treatment, and trials—come its new challenges, and clinical laboratories are doing what it takes and sharing how. Two plenary speakers at last year’s meeting of the Association for Molecular Pathology spoke of variant calling in the bioinformatic pipeline and validation, and of clinical reporting. Colin Pritchard, MD, PhD, of the University of Washington and one of the speakers, sees reporting a genomic sequencing assay as more like making a histologic diagnosis, which he calls craftwork, than reporting a sodium value. “That’s an idea that hasn’t really permeated yet,” he said.
In a separate AMP workshop, Brian Shirts, MD, PhD, presented an expanded view of clinical reporting in his talk on communicating genomic information to physicians. Dr. Shirts is working with two large collaborative groups funded by the National Human Genome Research Institute to improve reporting. “Our long-term goal is to influence providers of electronic health records and policymakers to improve the presentation of clinical genetic information,” Dr. Shirts, an assistant professor in the Department of Laboratory Medicine at the University of Washington, said in an interview.
“The EMR is not just an electronic version of the paper record,” Dr. Shirts said. “It should do something more than just be a file system for bunches of documents.”
Results of an exploratory survey of 17 medical institutions made clear that there is no consensus at this time about where genetic information should go in the electronic health record or how it should be displayed. Dr. Shirts described the situation as “chaos.” Recommendations have been made; figuring out how best to implement them will take considerable further work.
The three presenters spoke recently with CAP TODAY.

Dr. Berger
Next-generation sequencing is performed as highly focused “hotspot” panels, large gene panels, and whole exome sequencing. (See “In next-gen sequencing, panel versus exome,” CAP TODAY, January 2016, page 1). Michael F. Berger, PhD, and his colleagues in the Department of Pathology at Memorial Sloan Kettering Cancer Center have settled on a midsize cancer gene panel that detects 5,770 protein coding regions of 410 genes and 46 introns, which they call Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets, or MSK-IMPACT (Cheng DT, et al. J Mol Diagn. 2015;17:251–264). He reported in the plenary session the results of a large clinical evaluation of the panel.
“Hotspot panels are appropriate for some institutions only doing clinical applications,” Dr. Berger told CAP TODAY. Their advantages are shorter TAT and smaller amount of DNA required. They could be less costly because they would require smaller sequencers.
“It all depends on your throughput,” said Dr. Berger, associate director of the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. He and colleagues perform panels now for more than 500 patients per month; the goal is 10,000 to 15,000 cases per year. “With many samples like we are doing and investing in larger sequencers, the cost difference [between a hotspot panel and a larger gene panel] is not so great. And we can generate a lot more data per sample at a lower cost per base pair.”
The midsize cancer gene panel is scalable and more easily identifies copy number alterations and rearrangements because it involves exon capture by hybridization, not PCR amplification. And the panel’s greater depth of coverage gives them greater power to detect low frequency and subclonal mutations.
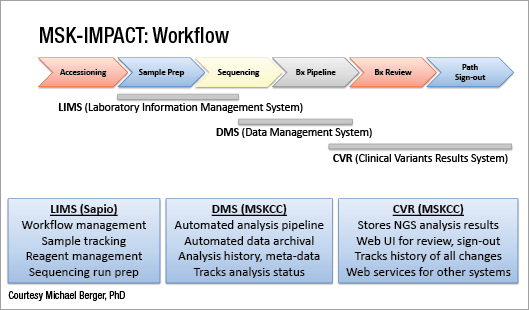
To validate MSK-IMPACT, Dr. Berger’s laboratory adhered to New York State Department of Health regulations. They observed each variant in 10 positive control samples, using hundreds of samples that had been sequenced in other clinical tests in their laboratory. In addition, they reported results across many runs and technologists.
In the MSK-IMPACT Clinical Sequencing Initiative, the laboratory focused on patients with recurrent or metastatic cancer because they are the patients most likely to be helped. Results were used to provide diagnostic and prognostic information, to select targeted therapies, and to screen patients for clinical trial eligibility.
The billable cancers were lung, colon, melanoma, thyroid, and gastrointestinal stromal tumors, all of which made up much less than half of the samples investigated. “The majority fell into the ‘nonbillable’ category,” Dr. Berger said. “We are absorbing the cost to find out more about patient profiles.”
A meaningful result was obtained on 84 percent of about 8,000 samples. Among these 6,800 cases, breast, colorectal, and non-small cell lung cancers were the most frequent. In all, they sequenced more than 60 general tumor types and more than 300 specific tumor types.
A median of four mutations were found per sample (mean was seven), which means that some samples had many mutations. “When you have so many mutations, most are passengers,” Dr. Berger said. In these instances, prior cases can offer guidance. At MSKCC they use cBioPortal, a software tool developed in-house that can be accessed via any Internet browser and contains results from more than 8,000 cases. Noting which variants have been observed in many other cancer cases can make it easier to identify likely driver mutations.
Among the 6,800 successfully sequenced DNA samples, the most frequently altered cancer genes were TP53, TERT, KRAS, PIK3CA, and APC.
Dr. Berger addressed the conundrum of germline mutations. “We have avoided returning germline results” when seeking variants in cancer genes. “Initially,” he said, “intentional germline analysis was not performed. Only results found unintentionally during quality control were returned. We do a lot of careful QC for every run.”
“One thing we do is to look for evidence of tumor in the normal tissue. We look at copy number of normal diploid cells. Occasionally there might be an inherited copy number alteration. In those instances, because patients were signing a consent form, there was discussion if and how those unintentional germline findings should be returned.”
New York state approved MSK-IMPACT as a germline test in May 2015. From that time on, patients were able to opt in to simultaneous germline testing after viewing an instructional video and providing additional consent. Eighty-two genes in MSK-IMPACT are associated with cancer predisposition.
Anonymized analysis of the first 1,570 cases in which germline analysis was performed revealed that 13 percent had one (or more) pathogenic or likely pathogenic variant. What was unexpected was that only 81 of 198 (41 percent) presented in canonical tumor types. For example, only 28 of 52 (54 percent) of BRCA1/2 variants were found in breast, ovary, prostate, or pancreas cancer, which Dr. Berger calls “surprising and a little bit confusing.” These mutations presented also in patients with no family history.
While cBioPortal acts as a source of genomic information for clinicians, it can also work in the reverse direction. Clinicians can put relevant clinical information into the patient rec-ord, which will allow clinical and genomic correlations to be discovered. “Crowdsourcing among physicians at Memorial is hard to do,” Dr. Berger said. “This part of the project will require collaboration with other places.”
Another ongoing downstream study seeks outcomes data from MSK-IMPACT results. It aims to answer these questions:
- How many cases harbor actionable mutations?
- In how many cases is management changed based on molecular alterations?
- Do mutations correlate with outcomes or response to therapy?
- Are responses modulated by co-mutational patterns?
- Is tumor heterogeneity related to response?
Dr. Pritchard, associate director of UW Medicine’s clinical genetics and solid tumors laboratory, showed a slide in his talk of a Boeing 747 towing a child on a tricycle. The caption: “Too much power?” “Our ability to sequence the genome greatly outstrips our ability to understand genomics,” he said.
“Right now in the clinical space the sweet spot is for targeted panels that focus narrowly on a few hundred genes. With these panels you can maximize the quality of the data you get and still be able to interpret the gene variations you find within the scope of disease you are considering.”
Dr. Pritchard presented the pros and cons of buying versus building the software infrastructure necessary for next-gen sequencing. His group came down on the side of building. (See “IT staffing considerations for the NGS laboratory,” CAP TODAY, February 2016, page 77.)
To be able to detect all forms of variants, from single nucleotide variants to deletions and rearrangements, Dr. Pritchard recommends parallelization—running the data through several variant callers in parallel. “Variant callers can have different strengths,” he said. “Stacking multiple approaches increases sensitivity.” This is especially important for structural variants. As with all assays, however, increasing sensitivity lowers specificity. One approach is to use multiple variant callers with different performance characteristics in parallel along with sophisticated manual review to minimize reporting of false-positive results. “This has been very important and successful for us,” Dr. Pritchard said.
Using this strategy, he and his colleagues developed the pipeline UW-OncoPlex, which provides simultaneous deep-sequencing information, based on greater than 500× average coverage, for all classes of mutations in 262 clinically relevant genes (Pritchard CC, et al. J Mol Diagn. 2014;16:56–67). In validation studies, UW-OncoPlex correctly identified 129 of 130 known mutations.
When adding targets to the panel, revalidation is essential. As with the initial validation, it is important to use both dry and wet controls and manual and automated processes. “When thinking about revalidating a pipeline, you want to run both DNA samples with known certain variants and data sets that have previously been called with the validated version of the pipeline.” The results should be evaluated with automated processes and manual review by experts who are familiar with the data. “I still want to lay eyes on it,” Dr. Pritchard said of manual review.
With NGS, coverage varies across the genome. “We need ways to flag low-coverage areas,” he said. A level of unacceptable coverage needs to be defined. While a sample might have average coverage of several hundred times, local coverage at the exon level might be much lower in some regions.
For further accuracy, each laboratory should establish an internal variant database, Dr. Pritchard recommends. This is a database, maintained at the institution, in which all variants detected in that laboratory are stored, along with interpretations. “In talking to my colleagues it seems like people are doing this,” he said. “It is not published so much, but molecular pathologists recognize it as important.”
An “error profile” is also a crucial database to maintain. Dr. Pritchard’s group is running 10 large capture-based sequencing panels for cancer as well as assays for inherited diseases. “It is critical to have an error profile on each assay,” he said.
Also integral to any pipeline will be custom genotyping. As an example, Dr. Pritchard displayed a screen shot of essentially raw data showing the variant MSH2c.942+3A>T, a common pathogenic Lynch syndrome mutation often missed by conventional variant callers because it is at the end of a string of A’s in the genome. “All platforms have a tough time with variants at the end of a homopolymer run,” Dr. Pritchard said. So his group built in a custom feature that says, “Tell me how many T’s there are at this position no matter what.” That flags the area for director review.
“In general,” Dr. Pritchard suggests, “anything that is really clinically actionable you may want to flag for analytical review plus expert review.”
To illustrate that special analyses can be done with customized NGS, Dr. Pritchard showed analysis by NGS of specimens for microsatellite instability (Hempelmann JA, et al. J Mol Diagn. 2015;17:705–714; Salipante SJ, et al. Clin Chem. 2014;60:1192–1199). “Directly determined MSI with NGS may be better than with capillary electrophoresis,” he said. Microsatellite instability has traditionally been done with capillary electrophoresis plus fragment ana-lysis, and that method remains predominant. “I think increasingly it is being recognized that MSI can be very accurately determined with NGS,” he said. “However, the bioinformatics have to be done appropriately.”