
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from the University of Texas MD Anderson Cancer Center. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Roberto Ruiz-Cordero, MD
Jeanne M. Meis, MD
Russell R. Broaddus, MD, PhD
![]() January 2019—A 75-year-old woman with history of melanoma localized to the right forearm and status post excision six years prior presented with a two-month history of continuous left shoulder pain. She was managed initially with physical therapy and hydrocodone with no effect. Initial workup at an outside institution included an x-ray of the left shoulder (Fig. 1A) that showed a destructive lytic lesion involving the proximal aspect of the left humerus associated with a pathologic fracture. A subsequent MRI (Fig. 1D) showed a dichotomous tumor measuring approximately 7.5 cm that disrupted the posteromedial cortex of the humerus and extended into the soft tissues. The superior aspect of the tumor showed septal enhancement, suggestive of enchondroma, while the distal aspect of the lesion showed disorganized, heterogeneous enhancement and a large soft tissue mass extending from the medullary cavity. These imaging findings were suggestive of dedifferentiated chondrosarcoma. Prominent axillary lymph nodes were observed; however, no distant metastases were identified.
January 2019—A 75-year-old woman with history of melanoma localized to the right forearm and status post excision six years prior presented with a two-month history of continuous left shoulder pain. She was managed initially with physical therapy and hydrocodone with no effect. Initial workup at an outside institution included an x-ray of the left shoulder (Fig. 1A) that showed a destructive lytic lesion involving the proximal aspect of the left humerus associated with a pathologic fracture. A subsequent MRI (Fig. 1D) showed a dichotomous tumor measuring approximately 7.5 cm that disrupted the posteromedial cortex of the humerus and extended into the soft tissues. The superior aspect of the tumor showed septal enhancement, suggestive of enchondroma, while the distal aspect of the lesion showed disorganized, heterogeneous enhancement and a large soft tissue mass extending from the medullary cavity. These imaging findings were suggestive of dedifferentiated chondrosarcoma. Prominent axillary lymph nodes were observed; however, no distant metastases were identified.
The patient was referred to our institution for further management, where an x-ray followed by an ultrasound-guided core-needle biopsy (Figs. 1B and 1C) were performed prior to receipt of the outside MRI studies. The histologic sections showed sheets of large epithelioid cells with well-defined cytoplasmic margins with osteoid deposition and a minute fragment of atypical cartilage. The initial morphologic differential diagnosis included metastatic melanoma, metastatic breast carcinoma, and high-grade sarcoma including dedifferentiated chondrosarcoma. Immunohistochemistry analysis performed on formalin-fixed, paraffin-embedded tissue sections showed that the tumor cells were positive for SATB2 (Fig. 1C, middle panel) and p53 (Fig. 1C, bottom panel) and negative for MART-1, SOX9, SOX10, BRAF-V600E, S100 protein, pankeratin, and GATA3 (not shown). The Ki-67 proliferative index was approximately 90 percent. The presence of osteoid with SATB2 and p53 positivity raised the possibility of osteosarcoma, either as a primary diagnosis or as the dedifferentiated component of chondrosarcoma. Since preoperative chemotherapy is usually given in cases of osteosarcoma or undifferentiated sarcoma and not routinely in cases of dedifferentiated chondrosarcoma, the distinction was clinically important.
An initial preliminary diagnosis of high-grade malignant neoplasm, possibly dedifferentiated chondrosarcoma, was rendered and FFPE tissue was sent to the molecular diagnostics laboratory for DNA extraction and targeted next-generation sequencing. We performed mutational analysis on the Ion Torrent semiconductor-based sequencer (Thermo Fisher Scientific) using a 50 cancer-related gene custom panel. Mutations in the IDH2 (NM_002168.2(IDH2):c.516G>T p.R172S) and TP53 (NM_000546.5(TP53):c.535C>A p.H179N) genes were identified (Figs. 1G and 1H). No mutations in the BRAF gene were detected. The presence of the IDH2 mutation confirmed the initial radiologic, morphologic, and immunophenotypic findings of dedifferentiated chondrosarcoma; thus the patient was eligible to participate in a pan-IDH inhibitor clinical trial.
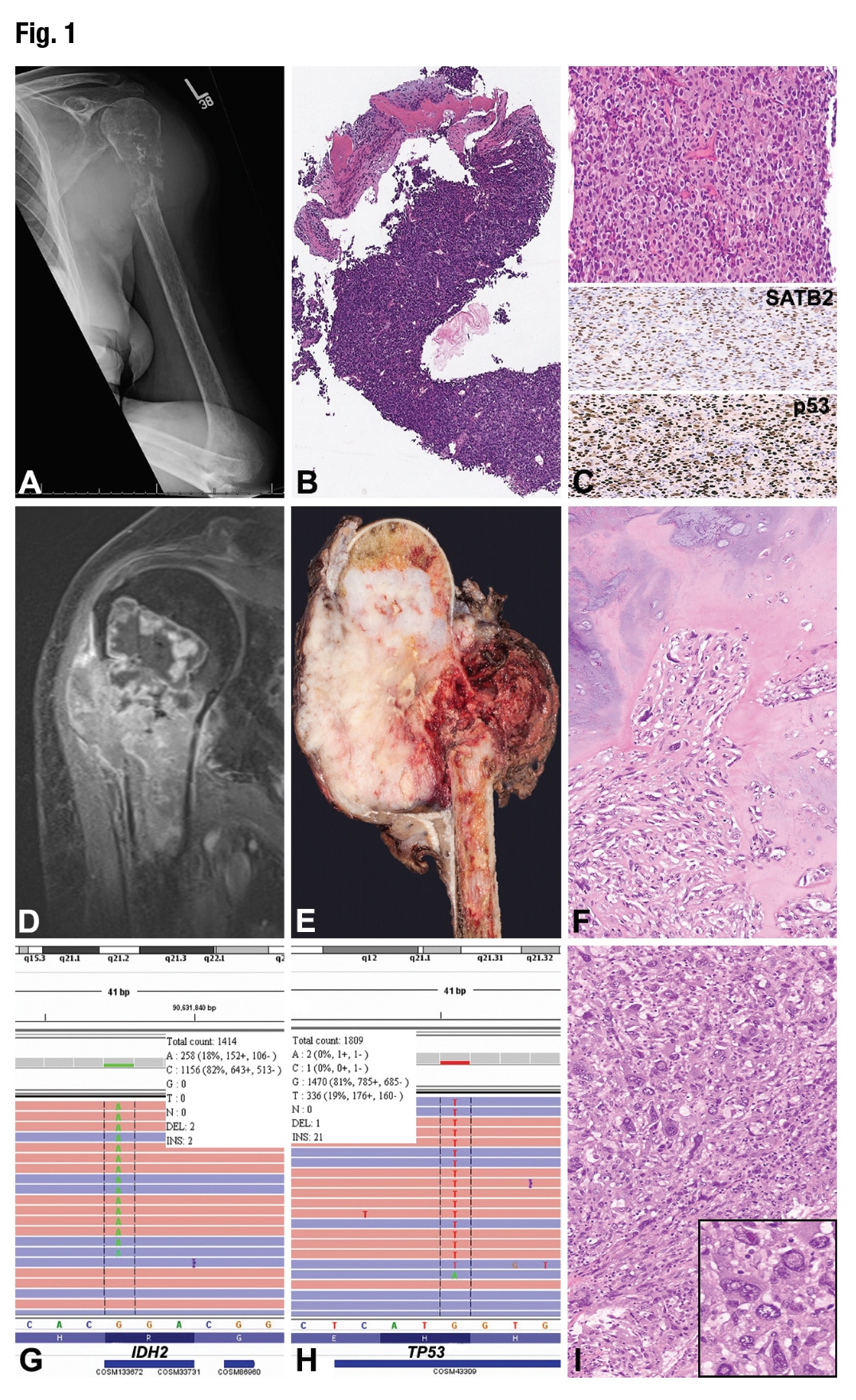 The patient underwent radical resection of the tumor (Fig. 1E) with endoprosthetic reconstruction of the left proximal humerus. Microscopic examination of the surgical specimen showed minor areas of conventional chondrosarcoma (Fig. 1F); however, the majority of the tumor was dedifferentiated with high-grade anaplastic features, including large and bizarre nuclei with pseudoinclusions, irregular nuclear contours, variable chromatin distribution, and eosinophilic cytoplasm (Fig. 1I, inset). The patient received the first cycle of IDH-inhibitor treatment, and radiation therapy was recommended. Unfortunately, the patient was lost to follow-up and died four months after surgery.
The patient underwent radical resection of the tumor (Fig. 1E) with endoprosthetic reconstruction of the left proximal humerus. Microscopic examination of the surgical specimen showed minor areas of conventional chondrosarcoma (Fig. 1F); however, the majority of the tumor was dedifferentiated with high-grade anaplastic features, including large and bizarre nuclei with pseudoinclusions, irregular nuclear contours, variable chromatin distribution, and eosinophilic cytoplasm (Fig. 1I, inset). The patient received the first cycle of IDH-inhibitor treatment, and radiation therapy was recommended. Unfortunately, the patient was lost to follow-up and died four months after surgery.
Mutations in the genes encoding the isocitrate dehydrogenase (IDH) 1/2 enzyme have been found in several tumors, including gliomas, some acute leukemias, cartilaginous tumors, thyroid carcinomas, cholangiocarcinoma, prostate cancer, paraganglioma, colorectal carcinoma, and melanoma.1 IDH2 catalyzes in the mitochondria the conversion of isocitrate to α-ketoglutarate, also known as 2-oxoglutarate (2OG), as part of the transformation of pyruvate into glutamate in the Krebs cycle,2 thereby playing a major role in cellular metabolism and energy production. Enzymes including histone methylases and other hydroxylases depend on 2OG to function properly.1 While somatic mutations of IDH1/2 were first described in gliomas,3 specific mutations in the arginine 172 residue of IDH2 have been reported in giant cell tumors of bone and cartilaginous tumors.4 Mutated IDH2 proteins acquire unique enzymatic activities that facilitate the production of oncometabolite D-2-hydroxyglutarate that inhibits 2OG-dependent enzymes.1 IDH2-R172S mutation has been found in 87 percent of enchondromas and 70 percent of spindle cell hemangiomas in association with Ollier disease and Maffucci syndrome5; however, IDH1/2 mutations can also be found in up to 87 percent of dedifferentiated chondrosarcomas and 30 percent of conventional chondrosarcomas.6 Moreover, IDH1/2 mutations in codons 132 or 172 are useful to discriminate between dedifferentiated chondrosarcoma and undifferentiated pleomorphic sarcoma of bone.6 The dedifferentiation process is complex and incompletely understood. Early reports identified similar genetic alterations in both components including loss of chromosome 13 and overexpression of p53, with the anaplastic component also showing severe aneuploidy, loss of heterozygosity at additional loci, and amplification and deletion of several other chromosomes.7 The TP53 missense mutation found in our case (H179N) has been reported in the Catalogue of Somatic Mutations in Cancer database as pathogenic in at least 27 different samples including primarily carcinomas of the gastrointestinal tract and one case of soft tissue sarcoma. Studies have shown that p53 mutant proteins have a longer half-life resulting in p53 IHC positivity in the majority of cases, as in our case, thereby making p53 IHC a good surrogate for TP53 mutation status.8
Limited sample size or quality can result in an equivocal microscopic diagnosis, and thus the identification by NGS of specific mutation(s) can aid in the final diagnosis and in managing the disease. In summary, this case demonstrates how the integration of clinical, radiological, pathological, and molecular data can help pathologists achieve the correct diagnosis, which is crucial for subsequent personalized therapy.
- Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18(20):5562–5571.
- Losman JA, Kaelin WG Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27(8):836–852.
- Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773.
- Liu X, Ogasawara S, Kaneko MK, et al. A novel monoclonal antibody SMab-2 recognizes endogenous IDH2-R172S of chondrosarcoma. Biochem Biophys Res Comm. 2015;459(4):636–642.
- Pansuriya TC, van Eijk R, d’Adamo P, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011;43(12):1256–1261.
- Chen S, Fritchie K, Wei S, et al. Diagnostic utility of IDH1/2 mutations to distinguish dedifferentiated chondrosarcoma from undifferentiated pleomorphic sarcoma of bone. Hum Pathol. 2017;65:239–246.
- Bovée JV, Cleton-Jansen AM, Rosenberg C, Taminiau AH, Cornelisse CJ, Hogendoorn PC. Molecular genetic characterization of both components of a dedifferentiated chondrosarcoma, with implications for its histogenesis. J Pathol. 1999;189(4):454–462.
- Murnyák B, Hortobágyi T. Immunohistochemical correlates of TP53 somatic mutations in cancer. Oncotarget. 2016;7(40):64910–64920.
Dr. Ruiz-Cordero is currently an assistant professor, Department of Pathology, University of California, San Francisco. However, the content of this report comes from, and the report was written at, the University of Texas MD Anderson Cancer Center, Houston, while he was a hematopathology fellow (and former molecular genetics pathology and cytopathology fellow). Dr. Meis is a professor of bone and soft tissue pathology, and Dr. Broaddus is a professor of gynecologic and molecular pathology—both in the Department of Pathology, University of Texas MD Anderson Cancer Center.
Test yourself: Here are three questions taken from the case report.
Answers are online now at www.amp.org/casereports and will be published next month in CAP TODAY.
1. Mutations in which of the following genes can help distinguish between dedifferentiated chondrosarcoma and undifferentiated pleomorphic sarcoma of bone?
a. MDM2
b. TP53
c. IDH2
d. PIK3CA
2. What molecule is isocitrate converted to by IDH1/2 enzymes?
a. D-2-hydroxyglutarate
b. 2-oxyglutarate, also known as α-ketoglutarate
c. Isocitrate
d. Citrate
e. Succinyl Co-A
3. What is the key oncometabolite produced as a result of gain of function of IDH2 due to mutations in the Arg140 or Arg172 residues?
a. D-2-hydroxyglutarate
b. 2-oxyglutarate, also known as α-ketoglutarate
c. Isocitrate
d. Citrate
e. Succinyl Co-A
Test yourself answers for December 2018 case report
In the December 2018 issue was a case report, “Discordant IHC/PCR test results for mismatch repair status in colorectal adenocarcinoma,” written by members of the AMP. Here are answers to the three “test yourself ” questions that followed that case report.
1. A recent report by Bartley, et al. (Cancer Prev Res [Phila]. 2012; 5[2]:320–327) estimates the rate of discordance between IHC and PCR-based assays for mismatch repair at approximately:
a. 0.1 percent
b. 2 percent
c. 10 percent
d. 20 percent
e. 55 percent
2. Why is it important to test for mismatch repair/microsatellite instability status of colorectal cancers?
a. It provides key information about tumor staging.
b. Patients with intact MMR have a better overall prognosis.
c. Patients with intact MMR should have routine extra-colonic cancer screenings.
d. It provides potential risk information for patients’ family members.
3. Which of the following is the correct interpretation of a tumor’s mismatch repair status with intact MMR protein expression by IHC but MSI-high by PCR?
a. Intact
b. Deficient
c. Indeterminate