 CAP TODAY Pathology/Laboratory Medicine/Laboratory Management
CAP TODAY Pathology/Laboratory Medicine/Laboratory Management

February 2016—Transfusion Medicine in the Hot Seat: A Case-Based Primer is a new book from CAP Press, due to be released next month. The editor, Karen Quillen, MD, MPH, and her five contributors wrote 26 cases grouped into three sections: Antibodies, Blood Components, and Complications. There are cases featuring anti-D in an Rh(D)-positive patient, blood management in obstetrics, acquired hemophilia A, hemolysis from platelet transfusion, hypotensive transfusion reaction, and much more. “Each chapter can be the springboard for weekly didactic sessions in transfusion medicine,” Dr. Quillen writes in her preface. Beginning on page 49 in this issue is her case titled “TTP or HUS?”
Dr. Quillen is a professor of pathology and laboratory medicine and a professor of medicine at Boston University School of Medicine. CAP TODAY asked her recently about the new book. Here is what she told us.
How did you select the cases for your book?
 The cases were selected to encompass the three key areas of transfusion medicine practice: interpreting antibody workups in patients who manifest RBC antibodies in pre-transfusion testing; selecting the appropriate blood components for different indications; and the recognition, diagnosis, and management of adverse reactions to transfusion. The cases are interesting—they’re not the average quick sign-outs—yet not zebras. Examples include the incompatible crossmatch as a classic anxiety-provoking situation, cryoprecipitate as a misunderstood and often misused component, and a spectrum of moderate to severe adverse reactions ranging from hypotension to hyperhemolysis. Molecular testing for Rh(D) variants (case 7) is a timely topic and the focus of a recent joint statement from the CAP and AABB and four other organizations.
The cases were selected to encompass the three key areas of transfusion medicine practice: interpreting antibody workups in patients who manifest RBC antibodies in pre-transfusion testing; selecting the appropriate blood components for different indications; and the recognition, diagnosis, and management of adverse reactions to transfusion. The cases are interesting—they’re not the average quick sign-outs—yet not zebras. Examples include the incompatible crossmatch as a classic anxiety-provoking situation, cryoprecipitate as a misunderstood and often misused component, and a spectrum of moderate to severe adverse reactions ranging from hypotension to hyperhemolysis. Molecular testing for Rh(D) variants (case 7) is a timely topic and the focus of a recent joint statement from the CAP and AABB and four other organizations.
You write in the preface that case-based teaching was the way you learned best and it is how you teach others. Exactly how do you do that today with pathology residents?
The concept of learning from “hot seat” cases will ring true with any pathology resident who’s been on blood bank call. I have never been a fan of didactic lectures because often the audience tunes out after the first few slides. My weekly teaching sessions with pathology residents are called tutorials to emphasize interactive discussion. They are almost never PowerPoint lectures—we start with “a case of the week,” which I select but a resident presents. It is typically a case encountered on the bench (antibody problems cases 1–8 in the book) or, more often, a call case where the resident on call was consulted about a problem, such as platelet transfusion refractoriness (case 9), or a question of plasmapheresis for thrombotic thrombocytopenic purpura (case 14). The chief resident, with my input, will select teaching material to accompany the case, such as a review article. An assigned resident will lead the discussion of the paper and around the topic. Sometimes the case and the topic of the day are different.
For what audience is your book most well suited?
I plan to use this book for residency teaching myself—the 26 cases will make a varied six-month transfusion medicine curriculum in clinical pathology. I think the book will be well received by pathology residents and hematology fellows for self-study, and by general pathologists with transfusion medicine responsibilities. The short case-based format makes it much easier to digest for those with busy schedules. Alumni residents who cover transfusion medicine within their general pathology practices have approached me for continuing education material; they have promised to get this book.
How did you select your contributors?
The contributors were selected to provide cases in areas that I don’t directly oversee, such as red blood cell genotyping and HLA testing. These tests are performed only in reference laboratories or large academic centers. From previous collaborations with the contributors, I know they all love to teach and like to write, although nobody particularly enjoys deadlines. It bodes well for the book that a couple of them have told me they had fun writing the “chaptettes.”
What would you say are the biggest challenges today in transfusion medicine?
The challenge of transfusion medicine practice is the need to make quick decisions in hot-seat situations. It’s stressful, particularly in the early months and years. Being able to communicate clearly and directly with a variety of staff—nurses, surgeons, laboratory technologists, internists—is a must. On a systemic level, there are many regulatory rules—CAP, Joint Commission, FDA, to name a few—and Part B reimbursement doesn’t really cover the time spent investigating complex cases, such as some of the serology or transfusion reaction cases illustrated in the book. On a societal level, well-trained medical technologists are worth their weight in gold, and payers need to fully reimburse the cost of blood so that newer technologies to continually enhance transfusion practice are affordable to hospitals.
TTP or HUS?
Case 14-1
A 70-year-old man is transferred from an outside hospital for shortness of breath and thrombocytopenia. He has had progressive shortness of breath with exertion for the past week. He denies chest pain or arm pain, fever, cough, orthopnea, or paroxysmal nocturnal dyspnea. He was diagnosed with a non–ST-elevation myocardial infarction at the outside hospital and new onset congestive heart failure the day prior to transfer. Coronary risk factors include hypertension and diabetes. He underwent pacemaker placement 15 years ago for bradycardia.
Past medical history is significant for untreated hepatitis C and benign prostatic hypertrophy. Medications include atorvastatin, finasteride, furosemide, labetalol, and metformin.
Vital signs: temperature 98.5°F, pulse 87, blood pressure 158/98 mmHg, respiratory rate 18, oxygen saturation 98%.
Laboratory results: WBC 8300/µL, hemoglobin 8.5 g/dL, hematocrit 28%, platelet count 31,000/µL, creatinine 1.5 mg/dL, LDH 742 U/L (reference ranges are shown with case 14-2). Peripheral blood smear review shows thrombocytopenia, large platelets, anisocytosis, poikilocytosis, many schistocytes, polychromasia, and occasional nucleated red cells.
Case 14-2
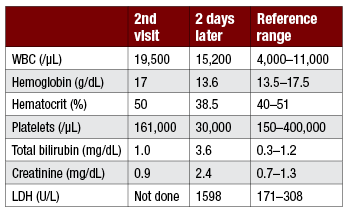 An 18-year-old college student presents to Student Health with bloody diarrhea and crampy abdominal pain with no obvious food precipitant. He is otherwise healthy. His roommates are well. Stool studies are sent. He is hydrated intravenously with 1 L normal saline and sent home. He returns 2 days later with worsening abdominal pain, continued bloody diarrhea, anorexia, and generalized malaise. Stool sample is positive for campylobacter antigen. Laboratory studies are on page 50. He is hydrated again and given a prescription for levofloxacin. He is able to take 1 or 2 doses only of the antibiotic, limited by nausea and anorexia. He presents to the emergency department for the third time 2 days after the second visit to Student Health with persistent gastrointestinal symptoms, generalized weakness, and dizziness. He has a low-grade fever to 100°F, blood pressure 95/70, pulse 120 regular, and looks fatigued but nontoxic. The state laboratory calls to report the detection of E coli O157:H7.
An 18-year-old college student presents to Student Health with bloody diarrhea and crampy abdominal pain with no obvious food precipitant. He is otherwise healthy. His roommates are well. Stool studies are sent. He is hydrated intravenously with 1 L normal saline and sent home. He returns 2 days later with worsening abdominal pain, continued bloody diarrhea, anorexia, and generalized malaise. Stool sample is positive for campylobacter antigen. Laboratory studies are on page 50. He is hydrated again and given a prescription for levofloxacin. He is able to take 1 or 2 doses only of the antibiotic, limited by nausea and anorexia. He presents to the emergency department for the third time 2 days after the second visit to Student Health with persistent gastrointestinal symptoms, generalized weakness, and dizziness. He has a low-grade fever to 100°F, blood pressure 95/70, pulse 120 regular, and looks fatigued but nontoxic. The state laboratory calls to report the detection of E coli O157:H7.
A representative blood smear, from a different patient, showing schistocytes (arrows) is shown in Figure 14-1.
Question: Which of these two patients should undergo emergent plasma exchange?
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura (TTP) for many years was a syndrome characterized by microangiopathic hemolytic anemia (MAHA) and thrombocytopenia, sometimes accompanied by renal insufficiency or neurologic abnormalities, in the absence of another cause for thrombotic microangiopathy (TMA).1,2 Conditions associated with secondary TMA include HIV infection; disseminated intravascular coagulation; systemic cancer; malignant hypertension; autoimmune disorders, particularly systemic lupus erythematosus and systemic sclerosis; hematopoietic stem cell transplantation; preeclampsia (HELLP syndrome); and certain drugs, such as quinine, calcineurin inhibitors, clopidogrel, and gemcitabine.3-5 The classic pentad—MAHA, thrombocytopenia, fever, and neurologic and renal compromise—occurs in less than 10% of patients ultimately diagnosed with TTP. 2
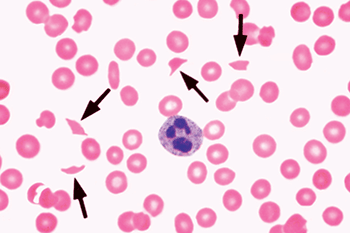
Figure 14-1. Representative blood smear showing schistocytes (arrows). From Glassy E, ed. Color Atlas of Hematology: An Illustrated Guide Based on Proficiency Testing. © 1998 College of American Pathologists.
Without plasma treatment, survival in TTP was only 10%. A landmark randomized trial in 1991 demonstrated that plasma exchange increased survival to 78%, compared with 63% survival in patients who received plasma infusion.6 Within a few years of this trial, deficiency in a von Willebrand factor-cleaving protease dubbed ADAMTS13 leading to unusually large von Willebrand factor multimers and platelet microthrombi in small vessels was discovered to be the pathogenesis of TTP. An ADAMTS13 level less than 10% of normal activity—in most cases attributed to an autoantibody or inhibitor against the protease—supports the clinical diagnosis of TTP and identifies almost all patients at risk for relapse after initial therapy, but half of TTP cases in the Oklahoma TTP-HUS registry did not have severe ADAMTS13 deficiency, and 20% of patients who had ADAMTS13 level less than 10% had secondary TMA.2
Despite the advances in our knowledge of TTP pathogenesis, the decision to initiate plasma exchange should be made in most cases on clinical grounds without waiting for the diagnostic test result, which frequently has a turnaround time of several days. On the other hand, plasma exchange is associated with some risk (detailed later), which makes it imperative that it not be initiated if secondary causes such as malignant hypertension or scleroderma crisis are overwhelmingly more likely to be the main culprit of the clinical picture. On rare occasions, pernicious anemia presents with severe anemia and thrombocytopenia, elevated lactate dehydrogenase (LDH) and bilirubin, and bizarre poikilocytes on the peripheral blood smear that could be mistaken for schistocytes.7 TTP is in many ways the classic “hot seat” case for the transfusion medicine physician.
TTP occurs predominantly in young to middle-aged adults, with a female preponderance. Neurologic abnormalities are present in two-thirds of patients, ranging from headache and confusion to focal symptoms/signs, seizure, stroke, and coma. Gastrointestinal symptoms also occur in two-thirds of patients. While pulmonary symptoms are rare, cardiac involvement is relatively common in TTP, manifested as chest pain, troponin elevation, or sudden death attributed to microthrombi in the cardiac conduction system. Median platelet count at presentation is 16,000/µL, hematocrit 25%, creatinine 1.1 mg/dL.2 Additional laboratory studies that should be part of the TTP workup include a direct Coombs test, INR/PTT and disseminated intravascular coagulation screen, HIV serology, ADAMTS13, baseline hepatitis B and C serologies, and, as clinically indicated in an older adult, a malignancy workup. Coagulopathy if present would suggest an alternative diagnosis or superimposed liver disease (not from TTP). A positive Coombs test would suggest autoimmune hemolytic anemia (case 4). Medication history is crucial, including dietary consumption of quinine (in tonic water) and illicit drugs such as cocaine.
The elderly patient in the first case manifests many clinical features of classic TTP; his age was the only outlier. The need for urgent plasma exchange was undisputed. Peripheral venous access was inadequate for apheresis. Although emergent bedside temporary apheresis catheter placement was possible, he appeared relatively stable, and the decision was made to transfuse fresh frozen plasma overnight (10 mL/kg or 3–4 units) with careful monitoring of his volume status, initiate corticosteroids, and await interventional radiology expertise. Indeed, his platelet count remained stable at 36,000/µL the next morning, apheresis catheter was placed uneventfully, and the first exchange was initiated within 18 hours of initial suspicion of TTP. Coagulation studies, direct Coombs test, and HIV serology were negative or normal. After the first plasma exchange, platelet count rose to 96,000/µL, although hemoglobin dropped to 6.8 g/dL, for which he was transfused 1 unit of blood. Platelet count normalized after the second plasma exchange. This rapidity of response was unexpected.
The median number of procedures to normalize platelet count in TTP is approximately 7 daily exchanges.2 Typically a longer course is needed in patients who have severe ADAMTS13 deficiency, which he turned out to have (ADAMTS13 <3%). After platelet count normalizes, an additional 1 or 2 sessions are performed as a “taper,” although this has not been rigorously studied. LDH improvement is rapid, but at the time of platelet count normalization, the LDH may still be slightly elevated (personal observation). While neurologic symptoms respond relatively quickly to plasma exchange, renal insufficiency and anemia take longer to improve. If platelet response is suboptimal or there is new neurologic abnormality, twice-daily plasma exchange or the addition of rituximab can be considered.8 The search for secondary causes of TMA should be intensified. Platelet transfusion in the absence of significant bleeding should be avoided, since it has been associated with worsening thrombosis.9 The superiority of cryosupernatant over plasma as replacement fluid for plasma exchange has not been proven; in theory, cryosupernatant does not contain von Willebrand factor (vWF), but there is no clinical evidence that vWF from normal blood donors exacerbates TTP.
Major complications occur in 25% of patients who undergo plasma exchange; these range from bacteremia, catheter obstruction, hemorrhage, pneumothorax or cardiac tamponade from catheter insertion, hypotension, and venous thrombosis, to anaphylaxis from plasma and death from sepsis or hemorrhage (3%).2 Less serious allergic reactions to plasma are relatively common despite the concurrent steroids that most patients are on and may hamper effective daily plasma therapy. Long-term transmission of infectious disease remains a concern, particularly in younger patients, despite modern nucleic acid testing for HIV, hepatitis C virus, and hepatitis B virus, although the advent of pathogen-inactivated plasma will reduce this risk further. Up to one-third of patients surviving the initial episode of TTP (with severe ADAMTS13 deficiency) may relapse, although the increasing use of rituximab during acute episodes may decrease the relapse rate.8 ADAMTS13 deficiency during remission is not clearly correlated with subsequent relapse and does not need to be monitored in the absence of symptoms or cytopenias.10 Prompt blood counts with any illness, particularly in the first year after recovery, are necessary to diagnose a TTP relapse.
Hemolytic uremic syndrome
Hemolytic uremic syndrome (HUS)—consisting of renal failure, MAHA, and thrombocytopenia—is more common in children. The sporadic form is diarrhea-associated or typical HUS. Abdominal pain and bloody diarrhea are manifestations of hemorrhagic enterocolitis caused by Shiga toxin-producing bacteria, most commonly E coli O157:H7 in the United States. This is also called Shiga toxin-mediated HUS (ST-HUS). Atypical HUS (aHUS) occurs when there is no diarrheal prodrome. Infection with Streptococcus pneumoniae in children is associated with a distinct form of aHUS characterized by T-activation and a positive direct antiglobulin test (case 8). Unless the etiology is pneumoccal-related, aHUS is more likely to be recurrent and has been recognized to be caused by defective complement regulation from genetic mutations of complement-regulatory genes (factors H and I are relatively more frequently implicated).1 Atypical HUS in adults can be distinguished from TTP by renal failure (three-quarters requiring dialysis, which is unheard of in TTP), less severe thrombocytopenia (median platelet count of 46,000 vs 12,000/µL), ADAMTS13 level >10%, and elevated biomarker of terminal complement activation (C5b-9).11 “Atypical” or complement-mediated HUS is treated with plasma infusion, plasma exchange, or an anticomplement agent such as eculizumab.1
ST-HUS is more common in children (median age of 2 years); management is supportive, and mortality is 3%. ST-HUS in adults is more severe with higher mortality (details later), popularized by large outbreaks in recent years traced to contaminated beef, spinach, and sprouts. Shiga toxin binds to CD77 on endothelial cells, renal mesangial cells, podocytes, and renal tubular cells. Cell apoptosis results from such binding through ribosomal inactivation. Shiga toxin is also prothrombotic by inducing endothelial secretion of vWF. In a series of 53 children infected with E coli O157:H7, thrombin generation and inhibition of fibrinolysis were more prominent in children who developed HUS and preceded renal injury.12 Severe abdominal pain and bloody diarrhea begin several days after consumption of contaminated food. Thrombocytopenia and MAHA develop as gastrointestinal symptoms resolve. Antibiotic administration for the diarrheal illness has emerged as a risk factor for subsequent development of HUS, presumably by causing the release of Shiga toxin from injured bacteria in the intestine. The antibiotics implicated in the study were trimethoprim-sulfamethoxazole and beta-lactam antibiotics to which the E coli strain was susceptible.13
There have been several large outbreaks of Shiga toxin-producing E coli infections in the last two decades. One was an E coli O157:H7 outbreak in 1996 in Scotland. Out of 200 confirmed cases of infection (median age 53 years), 28 met the criteria for HUS, on the average 1 week after the onset of gastrointestinal symptoms. Median age of adults with HUS was 71 years, about 20% required dialysis, just over half had neurologic features, and median platelet count was 121,000/µL.14 A decision was made to use plasma exchange in all the patients who could tolerate it, which came to 16 in 22 (73%). Mortality was 31% among the cohort who underwent plasma exchange for a median of 6 sessions, and overall mortality was 45%. In 2011, 3000 cases of Shiga toxin-producing E coli O104:H4 infection occurred in Germany and other European countries, with 491 confirmed cases of HUS. Median age was 46, neurologic symptoms were present in 69% of patients, and hemodialysis was required in 57%. The German Society of Nephrology published treatment suggestions that included plasma exchange with or without eculizumab for more severely affected patients and supportive care for the remainder (12%). Patients who underwent plasma exchange had more severe thrombocytopenia (30,000 vs 62,000/µL), renal failure, and neurologic symptoms. For example, 64% of patients who received concurrent plasma exchange and eculizumab had confusion, 53% had speech abnormalities, and 39% had seizures. Average treatment course was 6 sessions of plasma exchange, with or without eculizumab. Overall mortality was 4.1% in this series; the median age of patients who died was 70.15
The young man with ST-HUS in the second case did not have neurologic symptoms but did have moderately severe thrombocytopenia; nadir platelet count was 23,000/µL on hospital day 3. Renal insufficiency remained nonoliguric throughout except for 1 day of urine output of 450 mL (that could have been under-reporting); peak creatinine was 2.6 on hospital day 2. LDH was markedly elevated, peaking at 1999 U/L on hospital day 6, corresponding to a nadir hemoglobin of 6.5 g/dL (hematocrit 19%), at which point he was symptomatic and transfused. Decision to hold off on plasma exchange was based on daily assessments by a multidisciplinary team involving the intensivist, nephrology, hematology, and transfusion medicine consultants. ADAMTS13 result was reported from the reference laboratory at 89% on hospital day 4. He had transient ileus, which slowly improved. He was discharged on hospital day 10 feeling much better, with normal creatinine and platelet count, hemoglobin/hematocrit 7.2/22 (after a total of 2 units of blood transfused), and LDH 1074 U/L.
Conclusion
TTP is a “do not miss” diagnosis because emergent plasma exchange is lifesaving. Patients suspected to have TTP should be transferred to a facility where plasma exchange can be performed, and given plasma infusion until such procedure can be initiated. Baseline ADAMTS13 level should be obtained prior to plasma therapy. Many patients do not have neurologic compromise; renal insufficiency, if present at all, is mild. Secondary causes of TMA should be sought. Atypical HUS may be treated with plasma exchange (or infusion) or eculizumab. Management of ST-HUS in adults needs to be individualized. Although plasma exchange has been used in uncontrolled series, supportive care without plasma exchange in young adults who are monitored closely can make it possible to avoid the risks of plasma exchange. Efforts to develop specific anti-toxin therapy for Shiga toxin are ongoing.
[hr]Case objectives