 CAP TODAY Pathology/Laboratory Medicine/Laboratory Management
CAP TODAY Pathology/Laboratory Medicine/Laboratory Management

Erik R. Washburn, MD
Elizabeth E. Frauenhoffer, MD
Rina Kansal, MD
June 2016—CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Penn State Milton S. Hershey Medical Center and Penn State College of Medicine. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Mesenchymal neoplasms are typically characterized by gene fusions that occur due to chromosomal translocations, detection of which leads to a precise diagnosis.1 Among soft tissue sarcomas, these specific chromosomal translocations include the t(X;18)(p11;q11) for synovial sarcoma, t(11;22)(q24;q12) or t(21;22)(q22;q12) for Ewing tumor family (ES-PNET), and t(2;13)(q35;q14) or t(1;13)(p36;q14) for alveolar rhabdomyosarcomas. Molecular diagnostic tests have contributed immensely to accurate and specific diagnosis of soft tissue sarcomas,1,2 in part due to the limited utility of immunohistochemical stains.3 Synovial sarcoma (SS) is an aggressive sarcoma with a propensity for late local recurrence and metastasis. After rhabdomyosarcoma, SS is the second most common soft tissue sarcoma in children and adolescents. SS accounts for between five and 10 percent of all soft tissue sarcomas and most commonly occurs as a deep-seated tumor within the upper and lower extremities of older children and young adults.4 SS can display a variable degree of epithelial differentiation with a biphasic or monophasic pattern histologically. Greater than 90 percent of SS cases harbor a specific chromosomal translocation t(X;18)(p11;q11), leading to the formation of the SS18-SSX fusion gene, which can be identified definitively by molecular methods.1,2,5,6
We present a case of a 16-year-old female with a poorly differentiated synovial sarcoma, where the diagnosis was established by molecular diagnostic techniques, including reverse transcriptase polymerase chain reaction (RT-PCR) for detecting the SS18-SSX2 fusion transcript, and fluorescence in situ hybridization (FISH) for demonstrating the absence of EWSR1 gene rearrangement.
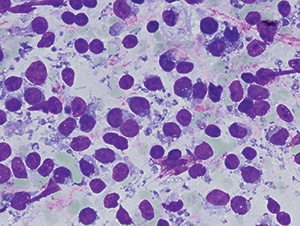
Fig. 1. Fine needle aspiration air-dried smear of right inguinal mass, showing dispersed monomorphous cells with scant cytoplasm, round nuclei with condensed chromatin, and inconspicuous nucleoli, consistent with small round blue cell tumor. Quik-Dip stain from Mercedes Medical, 1000× original magnification.
Case. A 16-year-old Hispanic female presented with a one-month history of proximal right thigh pain. Ultrasound showed a deep vein thrombosis in the right common femoral vein and a large complex mass in the right groin. Magnetic resonance imaging revealed an enhancing 9.3 × 8.9 × 7.2 cm mass of the right inguinal region, involving the adductor longus and adductor brevis musculature, with central necrosis. A 7.3 × 5.4 × 4.8 cm peripherally enhancing necrotic right common iliac lymph node was present, with no evidence of metastatic disease.
A CT-guided fine needle aspiration and core needle biopsy of the right inguinal mass demonstrated monotonous, overlapping, hyperchromatic ovoid spindle cell nuclei consistent with malignant small round blue cell tumor (Fig. 1). The core biopsy showed loosely cohesive groups of round-to-spindled cells with extensive necrosis (40 percent) within a fibrous background (Fig. 2). Tumor cells stained moderately for CD99, strongly for vimentin, with no staining for desmin, muscle specific actin, S100, CAM 5.2, or epithelial membrane antigen. Based on the histologic features and immunohistochemical staining, the differential diagnosis included extra-skeletal Ewing’s sarcoma and SS. Formalin-fixed, paraffin-embedded tumor tissue blocks were sent to Mayo Clinical Laboratories for RT-PCR and FISH studies, which were performed using previously described methods.7,8 The SS18-SSX2 fusion transcript, characteristic of SS, was detected by RT-PCR (Fig. 3). FISH showed absence of EWSR1 gene rearrangement, excluding Ewing’s sarcoma. The patient was given neoadjuvant chemoradiation therapy as per Children’s Oncology Group protocol ARST0332. A right external hemipelvectomy was performed, showing 94 percent tumor necrosis in the main tumor and 100 percent necrosis in the metastatic lymph node. At 17 months post therapy, the patient has no evidence of tumor recurrence.
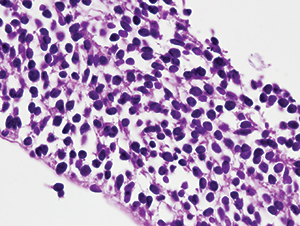
Fig. 2. Core needle biopsy of right inguinal mass, showing hypercellular sheet of monomorphous cells with hyperchromatic round nuclei, indistinct or absent nucleoli, and scant cytoplasm, within a loose, fibrous stroma, consistent with small round blue cell tumor. H&E stain, 400× original magnification.
Discussion. In this case, the histologic and cytomorphologic appearance of poorly differentiated SS closely resembled other sarcomas, in particular ES-PNET and rhabdomyosarcoma. By immunohistochemistry, negativity for the muscle specific markers excluded rhabdomyosarcoma. Ewing’s sarcoma could not be excluded, however, since CD99 and keratin can be expressed in both Ewing’s and SS.3,5
While most pediatric sarcomas may require a combination of neoadjuvant chemotherapy, radiation, surgery, and long-term follow-up, the specific protocols may differ depending on the type of sarcoma.9 Therefore, precise diagnosis, achieved by molecular or cytogenetic methods, is critical. The SS tumor-specific t(X;18)(p11;q11) translocation can be identified by conventional cytogenetic karyotypic analysis.10 While a cytogenetic analysis provides a global analysis of all chromosomes and can detect any additional secondary cytogenetic abnormalities, it requires a 1- to 2-cm3-sized fresh, viable, non-necrotic tumor sample, which is possible to obtain only from resection specimens and is not feasible from small needle core biopsies, as in this case. For formalin-fixed, paraffin-embedded tissues, a molecular cytogenetic assay such as FISH or a molecular method such as RT-PCR may be used to identify specific fusion genes or the fusion transcript, respectively.11,12 Both FISH and RT-PCR are designed to detect only a specific molecular genetic abnormality, without examining the remainder of the genome (complete chromosomes) in the analyzed tissues.