 CAP TODAY Pathology/Laboratory Medicine/Laboratory Management
CAP TODAY Pathology/Laboratory Medicine/Laboratory Management

Anne Ford
June 2018—For biomarker testing and tissue conservation, all roads lead to next-generation sequencing, says Boaz Kurtis, MD, laboratory and medical director of Cancer Genetics in Los Angeles.
Dr. Kurtis, speaking in a webinar on NGS in routine non-small cell lung cancer biomarker testing, said, “There’s no other technology platform out there that can provide the amount of data we need today or will need in the future.” If next-generation sequencing is performed today, he said, “we will be ready for what we can reasonably anticipate the future holds.”
As more oncogenic driver alterations have been uncovered, with targeted therapies to match, the need for data has evolved and guidelines have captured that need, Dr. Kurtis said in the webinar hosted in March by CAP TODAY and made possible by a special educational grant from Thermo Fisher.
The National Comprehensive Cancer Network, for example, advises broad molecular profiling so any possible therapy can be identified, whether off-label or under clinical investigation. “There’s an understanding, even by the NCCN,” which names only four genes (EGFR, ALK, BRAF, ROS1), “of what the trajectory of NGS testing is and why it’s important.”
The guideline of the CAP, International Association for the Study of Lung Cancer, and Association for Molecular Pathology “puts forward the same idea,” he said, “that we have reached the point where we need to start adding more genes and doing so is best done in labs that perform next-generation sequencing.” So there’s a shift, he added, not only to broaden the profiling but also “specific mention of or even bias toward a particular technology platform, in this case next-generation sequencing.”
Next-generation sequencing begins with extraction of genomic DNA from patient samples, Dr. Kurtis said as part of a refresher on NGS. “After doing so . . . there may be a step where we shear that genomic DNA into smaller fragments so those fragments can be accessible to some of the reagents, such as hybrid capture probes, that are used in next-generation sequencing panels. Once we have genomic DNA, with or without fragmentation, the next invariable step in the process is the ligation of adaptors to the ends of those fragments.”

Why ligate those adaptors? First, doing so allows the fragments to be bound to a solid surface (a requirement before the sequencing step). Second, “when a next-generation sequencing run is begun, it is begun on a single tube that combines DNA from multiple samples all mixed together,” Dr. Kurtis said. “And the only way to discriminate one sample from another is by including a barcode that allows you to essentially multiplex, within that one tube, numerous samples.
“The distal-most component would be to ligate the genomic DNA fragment to a solid surface,” he continued. “There might be a molecular barcode involved, and there’s also a sample barcode as the third color . . . So of course if you were to multiplex 10 samples into a tube, each one of the 10 samples would have a unique barcode, which is basically a piece of synthetic DNA. It might be perhaps eight nucleotides long, give or take, and it is essentially a key to indicate to which patient that piece of DNA belongs.”
After the ligation of the adaptors, the next step is to clean up the sample. “It’s essentially to enrich our sample for the genomic DNA of interest, specifically if it has ligated adaptors, and then to clean up and essentially discard everything else,” he said. Next is amplification, in which the user takes the genomic DNA of interest and allows it to reach a critical mass so that signals from that genomic DNA will be detectable in the sequencing step that follows.
After sequencing is the bioinformatic component, the purpose of which is to assemble the sequenced DNA (“it was broken up in fragments”) and align it to a referenced genome (a normal human genomic sequence in a healthy state). After alignment, “you identify what variants, if any, exist in your sample of interest,” and then analyze the importance of those variants.
Of the two widely available technology platforms in clinical use, the one that uses semiconductor sequencing is the Ion Torrent from Thermo Fisher. “Semiconductor sequencing essentially involves the addition of synthetic nucleotides to your genomic DNA and creating a phosphodiester bond,” he said. “One of the natural byproducts of the creation of a phosphodiester bond is the release of a hydrogen atom. So what semiconductor sequencing does is essentially exploit the release of a hydrogen atom.”
In doing so it detects changes in pH that result from the addition of a specific nucleotide. The sequencer will back calculate by determining what nucleotide was added that resulted in the change of pH. That series of events occurs over many cycles until a sequence is assembled for the fragment of interest.
The Illumina MiSeqDx uses sequencing by synthesis. “The technology here is quite different,” Dr. Kurtis said. “It detects light emitted by fluorophores. So every synthetic nucleotide added in an attempt to sequence the DNA is attached to a fluorophore” unique to that nucleotide. “Each one of the four nucleotides has one of four unique fluorophores.”
After a nucleotide with its bound fluorophore is added successfully to genomic DNA, a laser zaps the surface and the light that’s emitted has a unique wavelength detected by a camera inside the system. Once it’s read and the color is determined, the sequencer will back calculate what nucleotide was added when that fluorophore was detected. The location from which that light was emitted is recorded, and then a series of analyses allows the sequencer to also back calculate what the sequence was based on—both location on the flow cell and the wavelength and the color emitted from that location.
One of next-generation sequencing’s main benefits, Dr. Kurtis said, is its comprehensive nature: “You take one sample, you perform one assay on that sample, and you generate one report, and that report can, at least for many assays, provide you information related to all types of genomic alterations, including mutations, fusions, and copy number variations, which, for all intents and purposes, really means gene amplifications.”
The analytical sensitivity (the ability to detect a real variant versus background noise at a defined level of reliability) is comparable to, if not better than, existing molecular diagnostic technologies. “The point is that the next-generation sequencing doesn’t sacrifice anything,” he said. “In fact, it’s more often than not an enhancement in an ability to detect a variant that could be significant for the patient.”
For laboratories that order NGS tests, one of the first considerations is the panel itself. Large or small?
A larger panel can mean more potential matches for a therapy, yet is more likely to yield variants of uncertain significance. Smaller panels require shorter run times and so could theoretically provide shorter turnaround times. However, the wait time that can often be involved to maximize the efficiency of a run can negate the turnaround time advantage that a smaller panel might have. “And there are many labs out there that, for efficiency purposes, want to maximize the throughput of a next-generation sequencing run.” The interpretation component for larger panels is often longer, given their tendency to return many variants of unknown significance.
Larger panels require more tissue. And that is a very real practical challenge, Dr. Kurtis said, especially for the small diagnostic specimens that come from core needle biopsy procedures or fine needle aspirates. He pointed to data published in November 2017 showing that of 1,354 NSCLC samples, 76 percent were core needle biopsies (the remainder were FNAs and surgical resections), and of those core needle biopsies, 79 percent had more than 25 percent tumor content (Yu T, et al. J Thorac Oncol. 2017;12[11][suppl 2]:S1845).
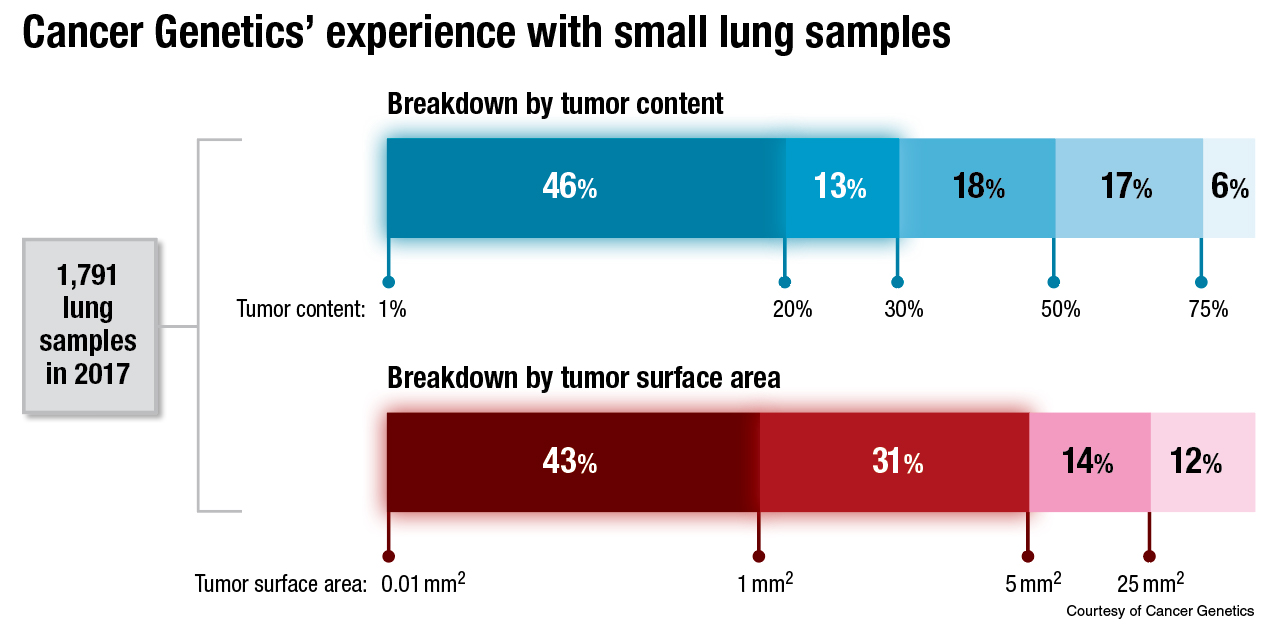
Dr. Kurtis and colleagues at Cancer Genetics have had a more concerning experience: Nearly half of 1,791 lung samples tested in 2017 had only between one and 20 percent tumor content. “Only the tumor cells will carry an alteration that you are going to be interested in,” he noted.
The thing to avoid, he continued, is having the normal DNA overwhelm the tumor DNA to the point that a mutation in the tumor DNA falls below the lower limit of detection. Enriching the tumor DNA within a sample is one way to avoid that. In Dr. Kurtis’ lab, that’s often done by dissection. “We have an entire team of trained folks here in our lab who perform dissection under a microscope, and in doing so, they literally scrape away tumor DNA and isolate it from surrounding normal [DNA], so as to bump up this percent content higher, as high as feasibly possible,” he said.
What about potential confounding variables? “Essentially, they’re all preanalytical variables, things that are out of our control,” he said, such as specimen type, processing, and storage duration and conditions, things that can “definitely impact the results of an NGS test.”
Tumor distribution is also a factor: “Is it distributed as an entire well-delineated block of tumor that could be easily separated by a dissection technique from surrounding normal tissue? Or is it disbursed in single cells or perhaps small clusters of cells haphazardly throughout the sample? That can often be the case in a cytology sample.” They see that frequently in cell blocks created from fluid samples. “You often have kind of discontinuous small pieces or small clusters of tumor, and pulling those together to form one mass of tumor DNA can often be a challenge, and often cannot even be accomplished with dissection technique.”
The subject of DNA and RNA integrity led him to speak about fluorometric quantitation. “When we talk about the trajectory of NGS testing, there’s actually another component to it,” he explained, pointing to a consensus recommendation of the CAP and Association for Molecular Pathology (Jennings LJ, et al. J Mol Diagn. 2017;19[3]:341–365). The guidelines for validating NGS-based oncology panels, “which even reached the point of how to quantitate the DNA and RNA,” he said, recommend using fluorescence-based technology rather than spectrophotometric methods. “In other words, there’s a recognition now that with NGS testing, we need to even standardize how that testing is prepared in the laboratory at the bench.”
The Thermo Fisher Oncomine Dx Target test used at Cancer Genetics, among other labs, is a 23-gene panel, with three genes (BRAF, ROS1, and EGFR) forming the companion diagnostic component of the assay. Analytically validated targets are KRAS, MET, and PIK3CA, with 18 additional targets such as AKT1 and CDK4. In contrast, the FoundationOne CDx panel encompasses 324 genes, with a variety of molecular alteration types detected. “So you can see how much more data a panel like this will yield as compared to a smaller panel,” with all the associated pros and cons, he said.
Dr. Kurtis closed with a snapshot of the current state of liquid biopsies and NGS. Quoting the most recent guideline of the CAP, IASLC, and AMP (Lindeman NI, et al. Arch Pathol Lab Med. 2018;142[3]:321–346), he said: “There is currently insufficient evidence to support the use of circulating plasma cfDNA molecular methods for establishing a primary diagnosis of lung adenocarcinoma . . . In some clinical settings in which tissue is limited and/or insufficient for molecular testing, physicians may use a cfDNA assay to identify EGFR mutations.”
In Dr. Kurtis’ view, “The panel that put these guidelines together basically hedged a little bit and said, in a certain scenario, you might want to consider using a liquid biopsy, but just for EGFR. And further, they added another caveat that if you believe your patient may be displaying clinical resistance to an EGFR-targeted tyrosine kinase inhibitor, you could consider using a cell-free DNA assay, the so-called liquid biopsy, to identify the resistance mutation such as EGFR T790M, the most widely known one.” Cell-free DNA may be preferred for patients unwilling or unable to undergo a biopsy at the time of progression, the guideline says.
“There’s some hedging here,” he said, “but there’s clearly a recognition that this is a component of molecular diagnostic testing that is going to evolve.” It’s a “hot space,” he added, “and it’s looking to grow even bigger.”
[hr]
Anne Ford is a writer in Evanston, Ill. The full webinar is at www.captodayonline.com.