 CAP TODAY Pathology/Laboratory Medicine/Laboratory Management
CAP TODAY Pathology/Laboratory Medicine/Laboratory Management

![]() September 2013—CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. Here, this month, is the third such case. (See the February 2013 issue for the first, on multilocus sequencing for rapid identification of molds, and last month’s issue for the second, on the importance of screening for Lynch syndrome in patients with endometrial cancer.) AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, treatment, and more. Case report No. 3 comes from Hartford Hospital, Connecticut Children’s Medical Center, and the University of Connecticut. (If you would like to submit a case report, please e-mail the AMP at amp@amp.org. For more information about the AMP, visit www.amp.org.)
September 2013—CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. Here, this month, is the third such case. (See the February 2013 issue for the first, on multilocus sequencing for rapid identification of molds, and last month’s issue for the second, on the importance of screening for Lynch syndrome in patients with endometrial cancer.) AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, treatment, and more. Case report No. 3 comes from Hartford Hospital, Connecticut Children’s Medical Center, and the University of Connecticut. (If you would like to submit a case report, please e-mail the AMP at amp@amp.org. For more information about the AMP, visit www.amp.org.)
Richard Danialan, DO; Arun Gopinath, MD
Michael Murphy, MD; Christine Rader, MD
Andrew Ricci Jr., MD; Jonathan Earle, MD
Zendee Elaba, MD
Abstract
An 18-month-old Hispanic female presented with an enlarging pigmented lesion on her leg. On excisional biopsy, histology showed an atypical melanocytic tumor with Spitzoid features. The differential diagnosis included Spitz nevus (SN), atypical Spitz tumor (AST), and Spitzoid malignant melanoma (SMM). Array comparative genomic hybridization (aCGH) studies were performed as a diagnostic aid and showed multiple chromosomal copy number aberrations, indicative of genomic instability and incompatible with a diagnosis of nevus. A diagnosis of SMM was made.
Introduction
Reliable distinction among SN, AST, and SMM is difficult to make solely on clinical and histopathological grounds because there is significant overlap of features. While several histopathologic criteria have been established in the literature to distinguish SN from AST and SMM,1,2 none have proven to be specific. As such, many borderline or equivocal melanocytic tumors are designated AST or “melanocytic tumor of uncertain malignant potential” (“MELTUMP”), with the notion that long-term followup would retrospectively categorize the lesions as either benign (no recurrence or regional metastasis) or malignant (distant metastasis or death).
Immunohistochemistry has shown limited utility in histologic differentiation. SN have initially been documented to show retained expression of p16, a tumor suppressor protein encoded by the CDKN2A gene on chromosome 9p21 while SMM and other melanomas exhibit loss of p16 protein expression.3 Recently, however, the dichotomous staining pattern of p16 in these lesions has been questioned, as 83 percent of SN and 79 percent of SMM expressed p16 in one particular study, demonstrating no significant difference.4 Helpful markers in the diagnosis of melanoma include MIB-1 and HMB-45. A MIB-1 proliferation index of greater than 10 percent has been shown to favor a diagnosis of SMM over SN, particularly at the deep end of the lesion.5 HMB-45 normally stains immature (type A) melanocytes with gradual loss of staining in the deep areas where mature (type C) melanocytes are located. SN are an exception as they may show diffuse HMB-45 staining. Melanomas show patchy HMB-45 staining throughout. Markers of melanocytic differentiation (Melan A, MITF, S100, Tyrosinase) do not distinguish benign from malignant melanocytes and are therefore not useful when the differential includes other melanocytic lesions.6
A number of molecular genetic techniques have been used as adjuncts in the diagnosis of atypical melanocytic lesions. Studies on the molecular profile of benign nevi, SN, and melanomas have illustrated certain chromosomal alterations characteristic of melanoma, such as gains in chromosomes 6p, 1q, 7p, 7q, 8q, 17q, 11q, and 20q, as well as losses in 9p, 9q, 10q, 10p, and 6q. Multicolor fluorescence in situ hybridization (FISH) assays evaluate four of these common numeric chromosomal aberrations (6p25, centromere 6, 6q23, and 11q13), allowing distinction between nevi and melanomas with a 95 percent specificity and 84 percent sensitivity in equivocal cases.7 aCGH examines the whole genome for numerical aberrations, with potential for enhanced sensitivity.8
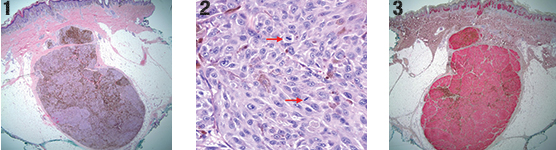
Fig 1. Atypical compound melanocytic proliferation with an expansile nodule extending into subcutis (100×, H&E).Fig 2. Atypical spindle to epithelioid melanocytes with mitotic figures (red arrows) (400×, H&E). Fig 3. Diffuse HMB-45 immunoreactivity (100×).
Patient case
An 18-month-old Hispanic female presented for what appeared to be an enlarging “dysplastic nevus” on the left lower leg, for which an excision was performed. Histology showed a compound proliferation of large epithelioid and fusiform melanocytes. The epidermal component showed irregular single and nested melanocytes with upward scatter and adnexal extension. No ulceration was present. A subjacent expansile nodule, composed of similar pigmented, epithelioid, non-maturing melanocytes with irregular nuclei, extended 8.5 mm from the superficial dermis to the subcutis (Fig. 1). Mitotic figures up to 3 per mm2 were identified within the deep dermal aspect (Fig. 2). Results of IHC studies were as follows: MIB-1 showed a proliferation index of approximately 10 percent in the dermal component; p16 showed diffuse cytoplasmic positivity with patchy loss of nuclear staining; melanocytes stained diffusely for HMB-45 with no difference in intensity from superficial to deep (Fig. 3). Due to the highly atypical histology and the patient’s young age, the formalin-fixed paraffin-embedded block was sent to the University of California, San Francisco, for aCGH studies, which revealed losses in chromosomes 1p, 8p, and 9, and gains in chromosomes 2 and 15q (Fig. 4). The identification of multiple chromosomal copy number aberrations indicates genomic instability, incompatible with interpretation as any type of melanocytic nevus. Following these findings, a diagnosis of childhood-type SMM was made. Currently, the patient is alive and well following a re-excision for close margins, which did not reveal any residual tumor.
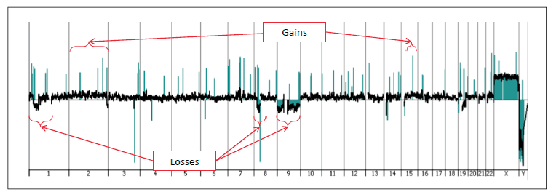
Fig 4. Tracing from aCGH analysis showing gains in chromosomes 2 and 15q, as well as losses in chromosomes 1p, 8p, and 9. The array (Agilent CGH microarray version 6.2.1, Agilent Technologies, Santa Clara, Calif.) was scanned using the Agilent microarray scanner G2505C and analyzed using Nexus Copy Number software 6.0 (BioDiscovery). (Courtesy of Tim McCalmont, MD, University of California, San Francisco.)
Discussion
In the case presented, the finding of multiple numeric chromosomal aberrations by aCGH was considered to represent genomic instability, inconsistent with a diagnosis of benign nevus. aCGH was initially employed in the study of melanomas by Bastian, et al.,8 in 1994, who described a number of chromosomal aberrations characteristic of melanoma, including loss of chromosome 9, which was the most common (81 percent), and was identified in the presented case. aCGH is a molecular cytogenetic assay that evaluates the entire genome for numerical chromosomal aberrations. In this assay, DNA from the patient’s test sample and normal human DNA are differentially labeled with fluorophores and hybridized to thousands of probes in both coding and non-coding regions in the human genome. A ratio of fluorescence between the tumor test sample and normal reference sample is obtained to determine genetic copy variations.9,10 A virtual karyogram is then compiled, as illustrated in Fig. 4.
Advantages of this technique include the relatively small amount of DNA required (typically one microgram) and the fact that both fresh and formalin-fixed paraffin-embedded tissue can be studied, allowing for the retrospective study of patient samples. Disadvantages include the significant cost of the test, its labor intensiveness, and the inability to detect balanced translocations, which could be significant in tumorigenesis. As with many molecular diagnostic techniques, signal-to-noise ratio and copy number calls are highly dependent on purity of microdissected tumor relative to admixed non-tumor cells. These technical considerations may be important in case selection.9,10
The final diagnosis in this case was arrived at from informed interpretation of complex molecular cytogenetic data in a unique clinicopathologic context, and would not have been possible without some understanding of both.