 CAP TODAY Pathology/Laboratory Medicine/Laboratory Management
CAP TODAY Pathology/Laboratory Medicine/Laboratory Management

![]() CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. Here, this month, is the second such case. (See CAP TODAY, February 2013, for the first, on multilocus sequencing for rapid identification of molds.) AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, treatment, and more. Case report No. 2 comes from Dartmouth-Hitchcock Medical Center, Hanover, NH. (If you would like to submit a case report, please e-mail the AMP at amp@amp.org. For more information about the AMP, visit www.amp.org.)
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. Here, this month, is the second such case. (See CAP TODAY, February 2013, for the first, on multilocus sequencing for rapid identification of molds.) AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, treatment, and more. Case report No. 2 comes from Dartmouth-Hitchcock Medical Center, Hanover, NH. (If you would like to submit a case report, please e-mail the AMP at amp@amp.org. For more information about the AMP, visit www.amp.org.)
Erik G. Jenson, MD
Gregory J. Tsongalis, PhD
Laura J. Tafe, MD
Abstract
August 2013—Lynch syndrome (LS) is an autosomal dominant syndrome that predisposes patients to multiple malignancies. LS has traditionally been thought of as a colorectal-cancer-dominated syndrome; however, the incidence of endometrial cancer in women with LS actually exceeds that of colorectal cancer. Here we report a case of a woman with metachronous colorectal cancer and endometrial cancer, with the goal of increasing awareness of the need to screen endometrial cancer patients for LS. Identifying these patients is important not only for the patient but also for other family members who would benefit from genetic counseling and surveillance for LS-associated malignancies.
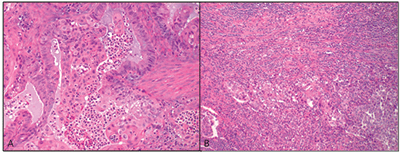
Fig. 1. The patient’s endometrial carcinoma showing (A) mucinous and squamous differentiation and (B) dense peri- and intratumoral lymphocytic infiltration (H&E, magnification 20×).
Introduction
Lynch syndrome, or hereditary non-polyposis colorectal cancer (HNPCC), is an autosomal dominant cancer susceptibility syndrome caused by germline mutations in one of four mismatch repair (MMR) genes: MLH1, MSH2, MSH6, and PMS2. The MMR proteins function as dimers (MLH1 with MSH2 and MSH6 with PMS2), and mutations in any one of these genes cause inactivation of the MMR system. This allows for the accumulation of unstable mismatched DNA in highly repeated microsatellite sequences, and gradually increasing instability with larger numbers of erroneous DNA segments (microsatellite instability, or MSI) and eventual gene expression alteration and subsequent carcinogenesis. Though Lynch syndrome was originally described as a familial predisposition to colorectal carcinomas, its association with carcinomas of noncolonic organs, such as endometrium, ovary, and stomach, among others, is now well recognized.
Lynch syndrome accounts for approximately two to three percent of CRC and 2.3 percent of endometrial cancers (EC), with an overall risk of developing CRC of 68 percent and EC of 62 percent in Lynch patients. However, when looking at the two genders separately, the risk of CRC for men is 83 percent versus 48 percent for women. Therefore, women with Lynch syndrome are at a substantially greater risk of developing EC than CRC, and in patients with metachronous cancers, 51 percent were diagnosed first with a primary gynecologic malignancy.1 Thus, while most pathologists and clinicians are aware of the association of CRC with LS, additional education on screening patients with endometrial cancer is needed.
Here we report a case of a woman with metachronous CRC and endometrial cancer who, despite a significant family history, was not evaluated for Lynch syndrome until her second primary tumor was identified. This case raises awareness of the association between LS and endometrial cancer and the modalities used to screen patients for LS, and the significance that identifying this syndrome can have on patients’ families.
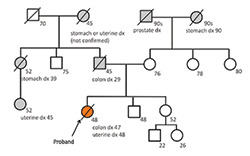
Fig. 2. The patient’s (Proband’s) family pedigree showing a number of relatives with Lynch syndrome-associated malignancies. The patient’s germline MLH1 mutation was most likely inherited from her father.
Patient case
A 48-year-old woman was referred to the gynecologic oncology outpatient clinic at our institution for evaluation of three weeks of vaginal bleeding. Per the patient, the vaginal bleeding was bright red and necessitated the use of pads. She had no further complaints and remained active, working at a local ski resort. Her gynecologic history was unremarkable with no pregnancies, normal Pap smears, and menopause five years prior. Exam revealed bright red blood in the vaginal vault, an exophytic lesion at the cervical os, and a large posterior uterine nodule appreciated on bimanual exam.
She had had an unremarkable personal health history until 10 months prior to this presentation. At that time, she presented to the ED for acute abdominal pain, ultimately determined to be a perforated colonic malignancy at the splenic flexure. A laparoscopic left hemicolectomy was performed at an outside institution and showed an acutely perforated, invasive, moderately differentiated mucinous adenocarcinoma (stage pT4 N0). Adjuvant chemotherapy (FOLFOX) was initiated after followup PETs demonstrated retroperitoneal nodal metastasis. The increasing adenopathy was followed via CT, which also showed “a mass arising in the endometrial cavity, infiltrative into the myometrium.” It had increased in size since it was first noted on prior imaging, prompting a referral to the gynecologic oncology clinic.
An endocervical biopsy showed endometrioid type adenocarcinoma (CK7 positive, CK20 and CDX2 negative). Subsequent TAH-BSO confirmed endometrial adenocarcinoma endometrioid type (FIGO grade II) with squamous and mucinous differentiation and foci of secretory change; the tumor was deeply invasive, involved the lower uterine segment and cervix, was associated with peri- and intratumoral lymphocytes, and had lymphovascular space invasion and metastasis to periaortic lymph nodes and one fallopian tube (pT3a N1 [IIIC]) (Fig.1).
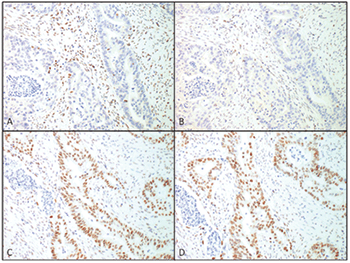
Fig. 3. Immunohistochemistry for the MMR proteins in this patient’s endometrial carcinoma showing complete loss of staining for MLH1 (A) and PMS2 (B) while staining is retained for MSH2 (C) and MSH6 (D) (magnification 20×).
Her family history was significant for multiple family members with endometrial, stomach, and colon cancers, including her father who was diagnosed with colon cancer at 29 and died of disease at 45 (Fig.2). Considering her family history and the diagnosis of two primary cancers, the question of Lynch syndrome was raised. In addition to routine H&E staining, immunohistochemistry was performed for DNA mismatch repair proteins MLH1, MSH2, MSH6, and PMS2. These stains showed a loss of MLH1 and PMS2 nuclear staining in tumor cells while MSH2 and MSH6 staining remained intact (Fig.3). Although most tumors exhibiting loss of MLH1 are associated with gene silencing through sporadic promoter methylation, this patient’s strong familial history suggested a possible germline mutation and further genetic testing was indicated. Gene sequencing performed at Myriad Laboratories revealed a MLH1 p.E13X (c.37G>T) deleterious nonsense mutation, confirming the diagnosis of Lynch syndrome likely inherited from her father.
Despite chemotherapy, the patient’s two metastatic diseases continued to spread rapidly and she died due to overwhelming tumor burden only a year after her initial presentation to the ED. Though the patient had no children, she had two siblings, a niece and nephew, and several cousins who will receive appropriate genetic counseling and subsequently be tested for this familial germline mutation.